|
2026-03-14
|
Bug Fix
|
fix(pipeline): Fix 11 NULL targets — DE condition, ID matching, consensus type
|
41
|
146
|
47
|
Other, R Source
|
|
2026-03-14
|
Bug Fix
|
fix(cachix): Remove –watch-mode auto flag (already default)
|
1
|
1
|
1
|
Other
|
|
2026-03-14
|
Bug Fix
|
fix(pipeline): Fix 3 NULL-target bugs, auto-generate package.nix (#93)
|
87
|
235
|
80
|
Config, Docs, Other, R Source
|
|
2026-03-14
|
Bug Fix
|
fix(nix): Fix cachix signing key, rebuild Bioconductor-dependent targets
|
2
|
0
|
0
|
Other
|
|
2026-03-14
|
New Feature
|
feat(captions): Add dynamic captions to 34 table/plot targets
|
22
|
579
|
89
|
Other, R Source
|
|
2026-03-14
|
Bug Fix
|
fix(vignettes): Enforce zero-computation rule — 22 violations → 0
|
32
|
360
|
764
|
Other, R Source, Vignettes
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Convert kable RDS to data.frames, fix telemetry eval guards
|
18
|
8
|
2
|
Other, Vignettes
|
|
2026-03-13
|
Bug Fix
|
fix(ci): Save data frames (not DT widgets) to RDS for Nix portability
|
3
|
0
|
0
|
Other
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Use Quarto #| eval syntax for pkgdown-banner chunks
|
11
|
44
|
11
|
Vignettes
|
|
2026-03-13
|
Refactoring
|
refactor(targets): Move Bioconductor packages to per-target declarations
|
11
|
35
|
17
|
Other, R Source, Vignettes
|
|
2026-03-13
|
New Feature
|
feat(vignettes): Add code provenance, kable→DT conversion, caption compliance
|
35
|
1004
|
437
|
CI/CD, Other, R Source, Vignettes
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Skip NULL RDS in safe_tar_read, return invisible(NULL)
|
11
|
22
|
22
|
Vignettes
|
|
2026-03-13
|
Bug Fix
|
fix(glossary): Prevent double DT::datatable() wrapping in glossary-table chunk
|
1
|
3
|
1
|
Vignettes
|
|
2026-03-13
|
CI/CD
|
ci: Show quarto errors with quiet=FALSE, render individual vignettes in diagnostic
|
1
|
20
|
6
|
CI/CD
|
|
2026-03-13
|
CI/CD
|
ci: Add verbose quarto error diagnostics on build failure
|
1
|
14
|
1
|
CI/CD
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Strip Nix paths from DT widgets, auto-wrap data frames
|
25
|
66
|
28
|
CI/CD, Other, Vignettes
|
|
2026-03-13
|
CI/CD
|
ci: Add diagnostic quarto render step to debug build failure
|
1
|
17
|
0
|
CI/CD
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Revert safe_tar_read placeholder, guard gene-report
|
11
|
12
|
56
|
Vignettes
|
|
2026-03-13
|
Maintenance
|
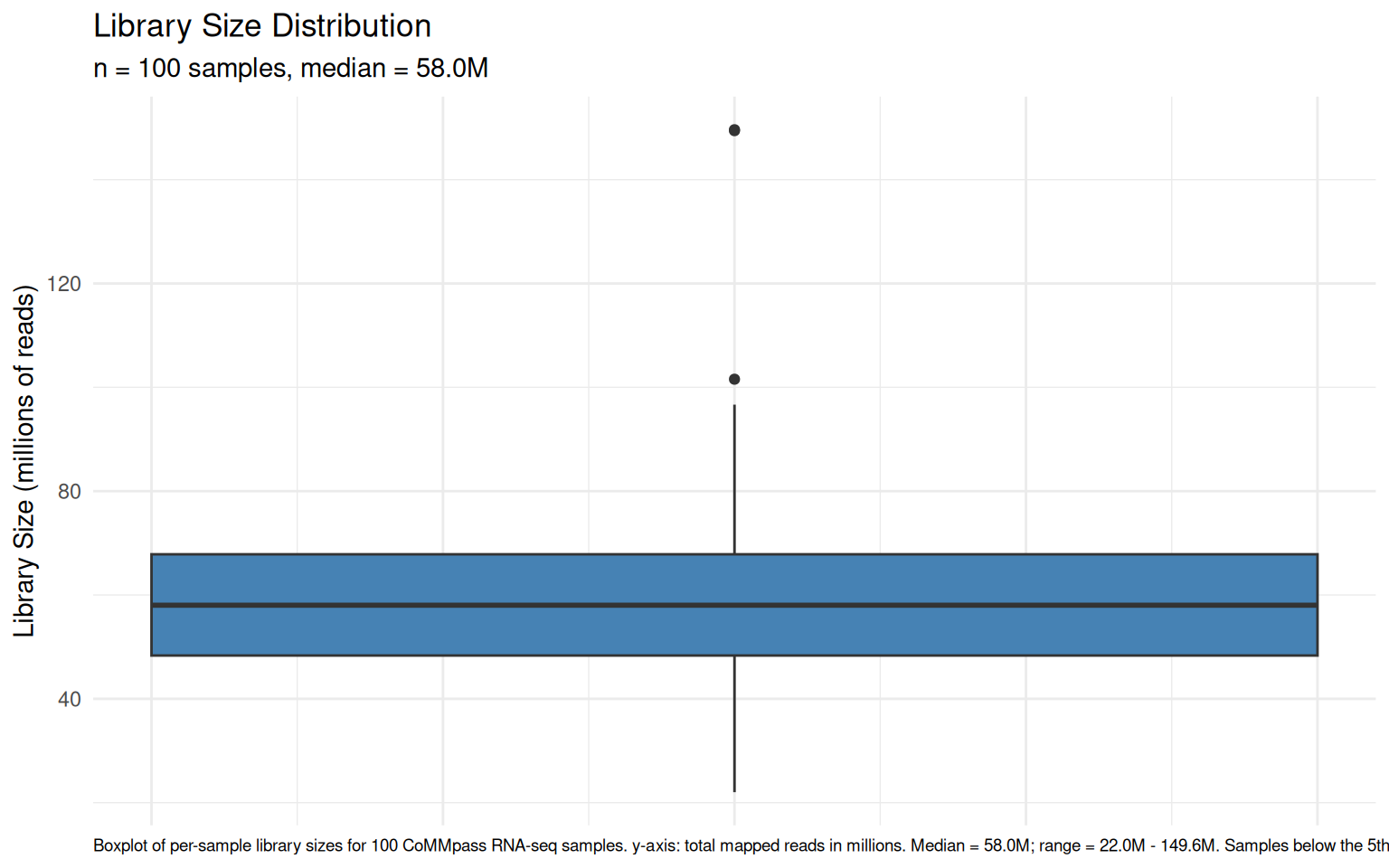
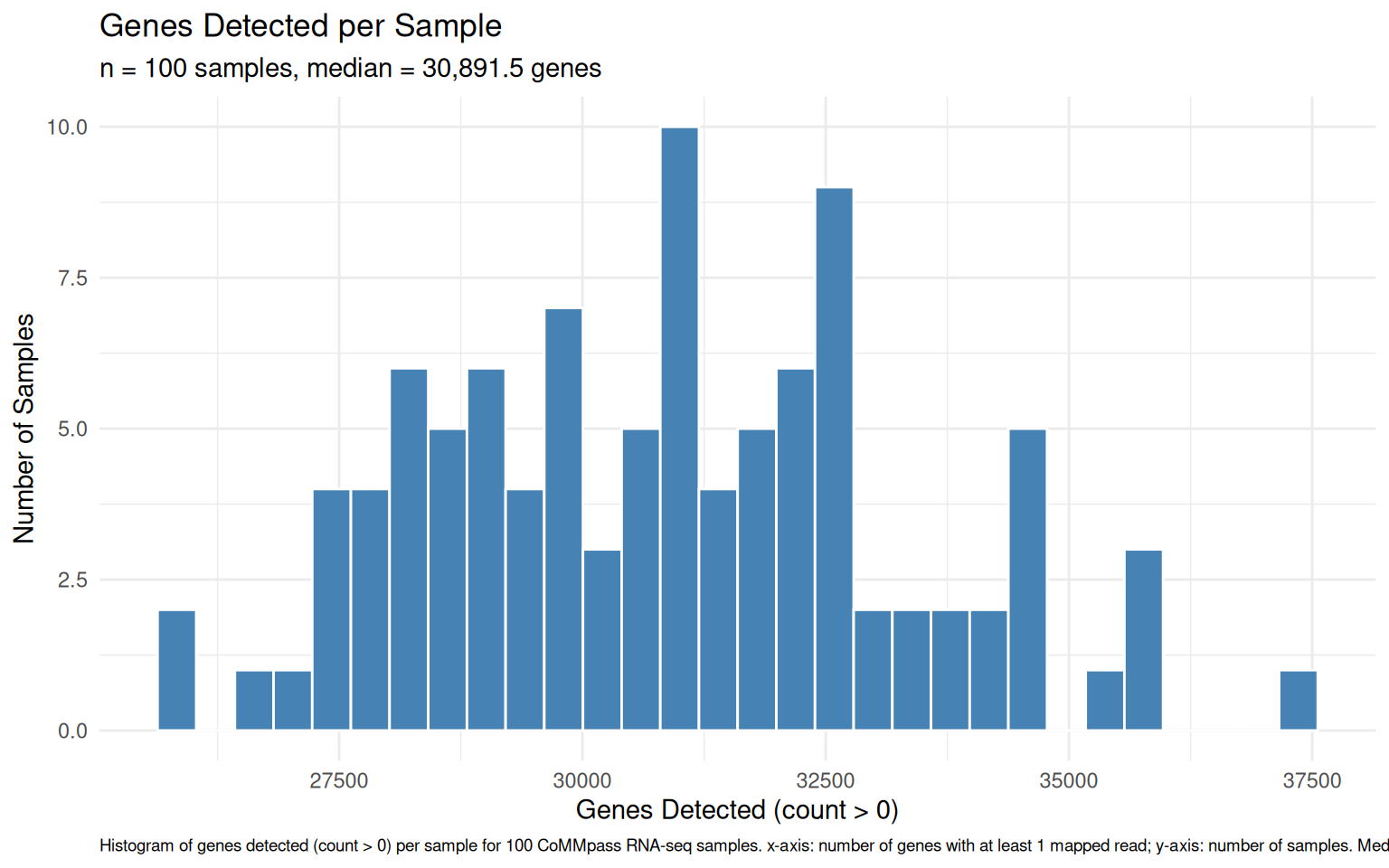
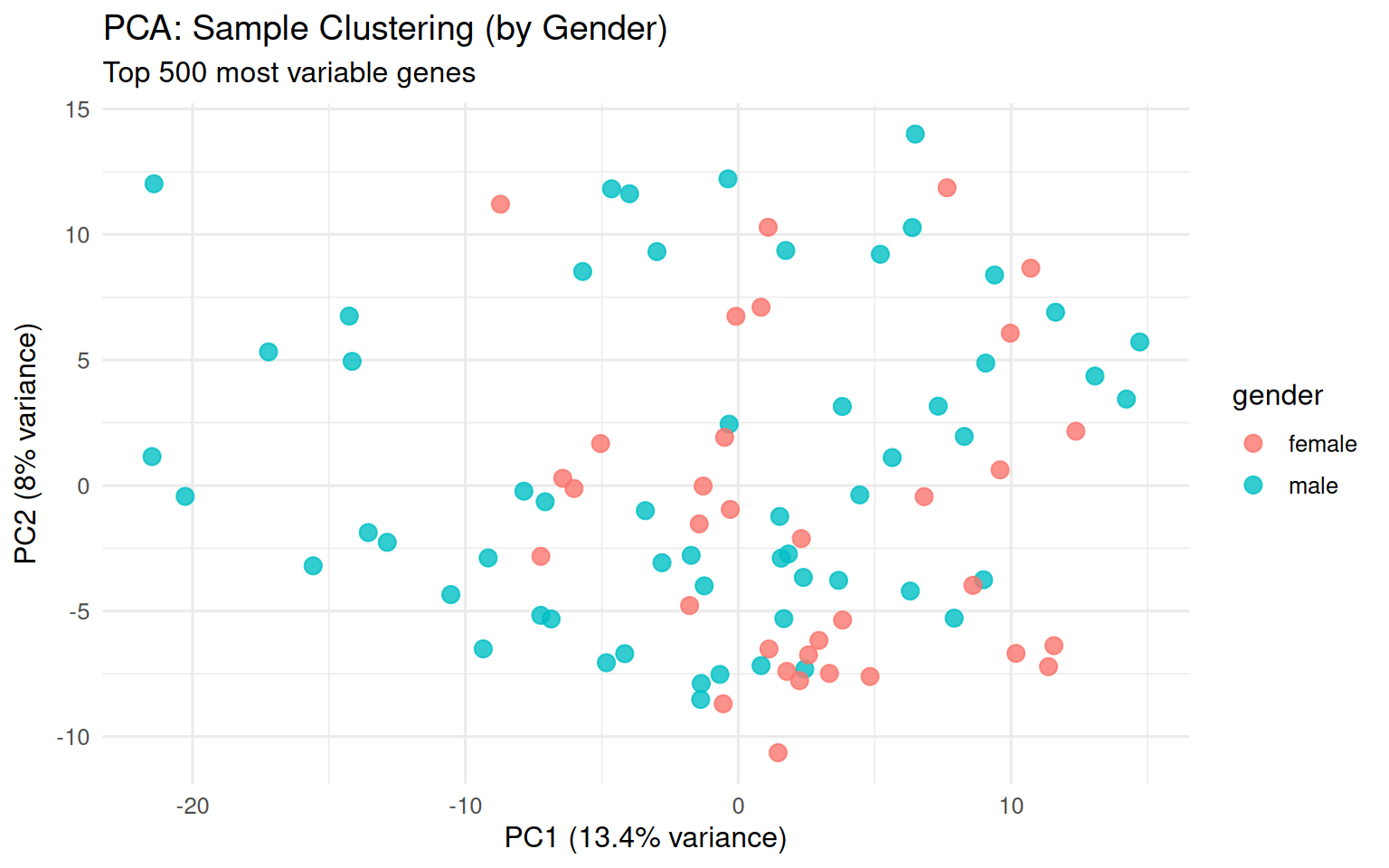
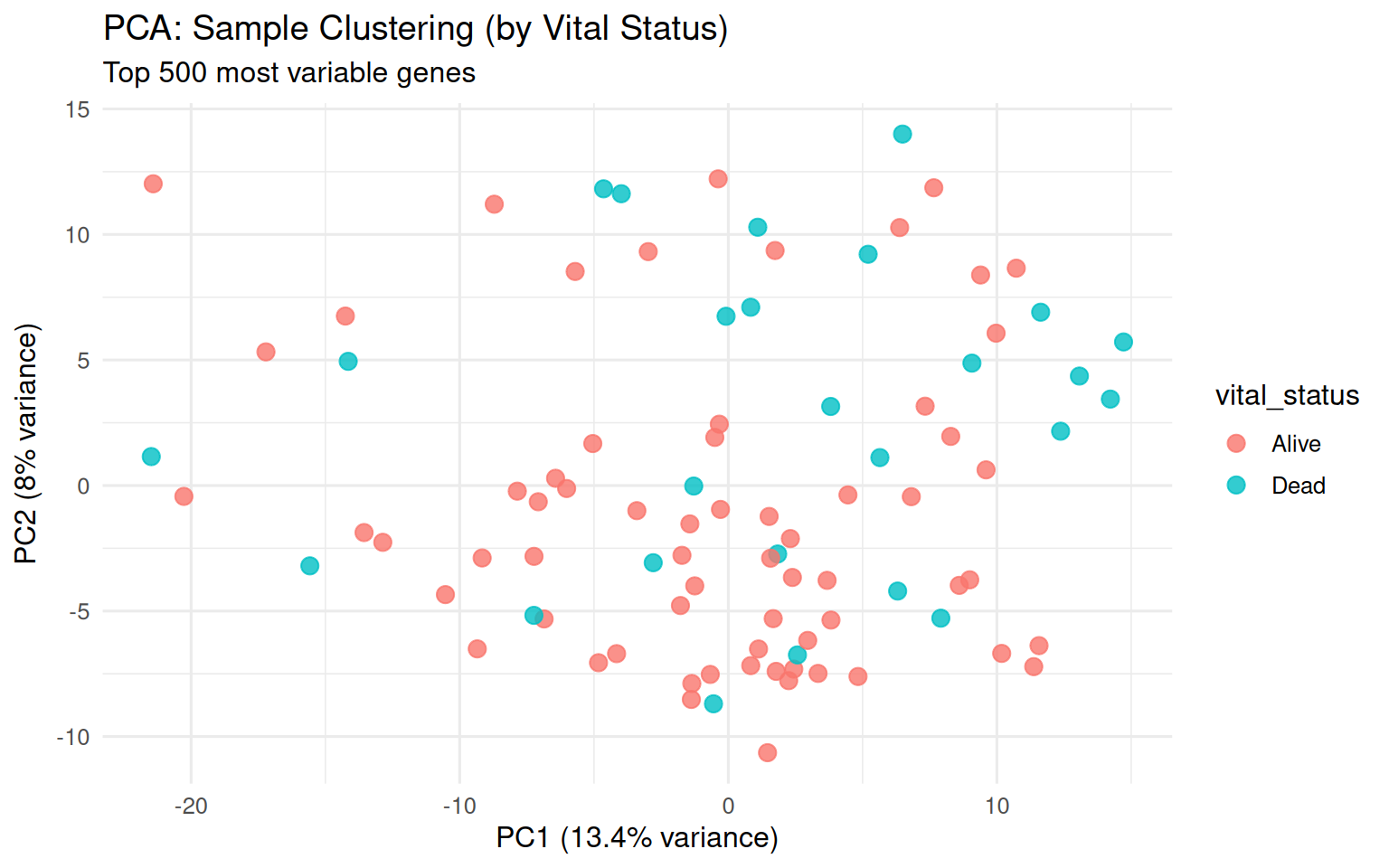
chore: Export vig_count_distribution_plot as ggplot RDS (513KB)
|
1
|
0
|
0
|
Other
|
|
2026-03-13
|
Bug Fix
|
fix(vignettes): Enable code eval in CI with RDS fallback
|
80
|
113
|
74
|
CI/CD, Other, R Source, Vignettes
|